发布日期:2026-05-24 来源:湖南医聊
5月23日是肝豆状核变性关爱日,今天中信湘雅生殖与遗传专科医院就来聊聊这个极具“杀伤力”的病。值得一提的是,在长沙市民生项目第一期的统计中,该病排名前十。
一、什么是肝豆状核变性?
肝豆状核变性,医学上称为Wilson病(Wilson Disease,简称WD),是一种常染色体隐性遗传的铜代谢障碍疾病。它的名字听起来复杂,但核心问题其实很简单——身体里的“铜”排不出去了。
正常人摄入的铜会通过胆汁等途径排出体外,而肝豆状核变性患者由于体内负责铜转运的ATP7B基因发生了突变,导致铜无法正常排出。这些“无处可去”的铜逐渐在肝脏、大脑、眼睛、肾脏等器官中堆积,最终引发“铜中毒”。
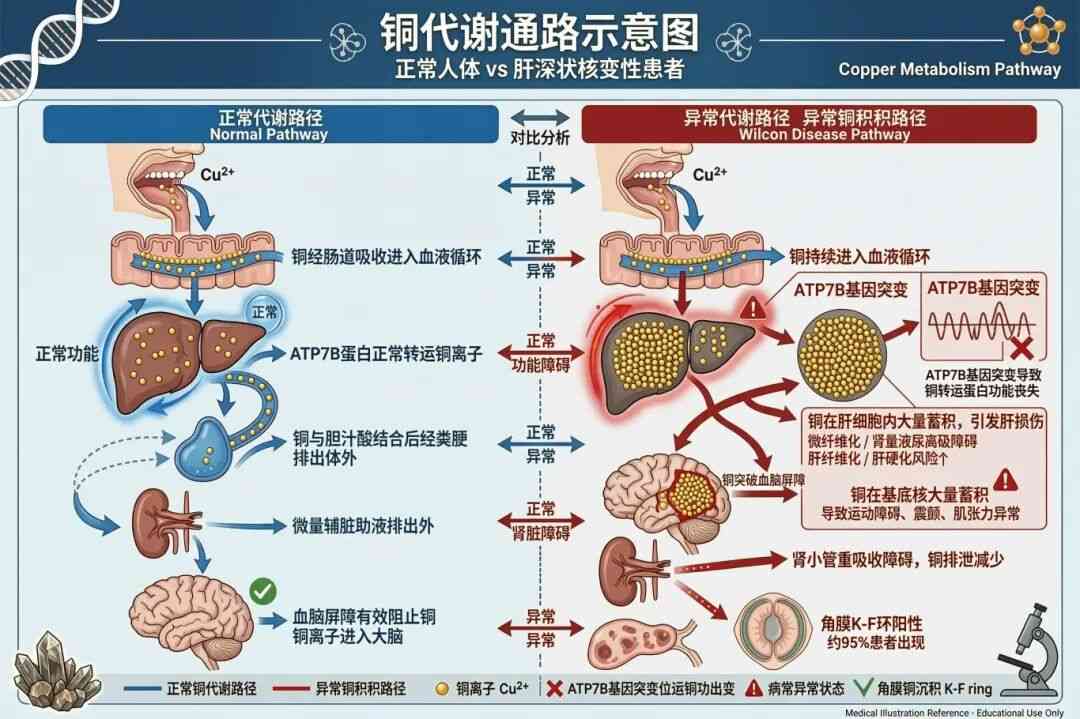
(图源:AI生成)
二、最“善变”的临床表现为什么容易被误诊?
肝豆状核变性有一个让所有医生头疼的特点:临床表现极其多样,早期症状不典型,极易被误诊。很多患者辗转多个科室才最终确诊。
典型的误诊案例:
1、因肝功能异常被当作肝炎治疗,反复无效后才查出WD;
2、因突发不自主运动被误诊为舞蹈病,最终通过铜代谢检查才确诊;
3、因行为异常、思维混乱被诊断为精神分裂症,长期治疗无效后才发现是WD。
为什么容易误诊?
中信湘雅生殖与遗传专科医院遗传中心罗俊杰医生表示,肝豆状核变性之所以成为误诊的“重灾区”,原因在于其临床表现的高度异质性和疾病进程的隐匿性。铜离子可在肝脏、大脑、肾脏、骨骼等不同器官沉积,导致首发症状千差万别,且早期症状往往不典型。
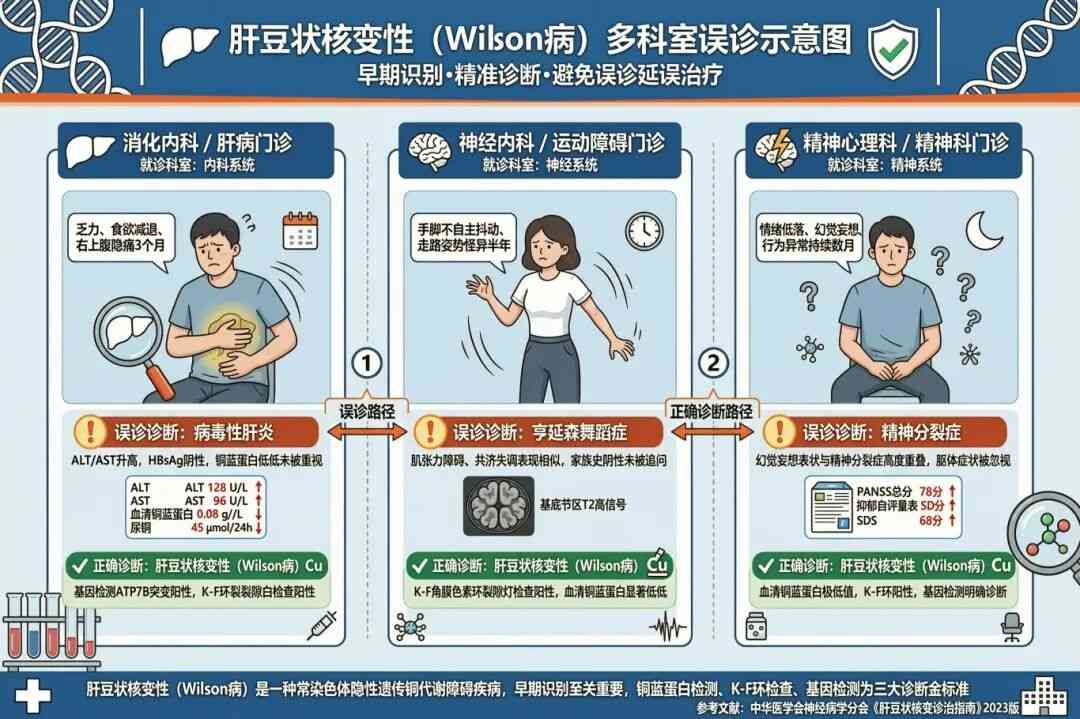
(图源:AI生成)
中信湘雅生殖与遗传专科医院遗传中心罗俊杰医生提醒:对于任何不明原因的肝脏损伤、神经系统症状或精神行为异常,尤其是有家族史的患者,都应警惕肝豆状核变性的可能。
三、诊断与治疗:早发现是关键
1、如何诊断?
目前临床使用最广泛的是Leipzig评分系统,综合K-F环、血清铜蓝蛋白水平、24小时尿铜量、肝组织铜定量、基因检测等多项指标打分,总分≥4分即可确诊。其中,通过基因检测检出ATP7B复合杂合/纯合致病性变异,被认为是诊断的金标准。
2、能治吗?——可治,但需终身管理
好消息是,肝豆状核变性是少数可以治疗和控制的遗传代谢病之一。若能在早期得到规范治疗,大部分患儿可获得正常寿命。
治疗的核心是“排铜”:一方面通过青霉胺、曲恩汀等螯合剂促进体内多余的铜排出,另一方面通过锌剂等减少肠道对铜的吸收。
同时,患者需要长期坚持低铜饮食,限制动物肝脏、贝类海鲜、坚果、巧克力等含铜量较高食物的摄入。需要强调的是,治疗是终身的,不可随意停药。
四、三代试管婴儿技术:从源头阻断遗传
肝豆状核变性是常染色体隐性遗传病,患者的父母为携带者,每次自然受孕,后代将有25%的概率患病,50%的概率成为携带者,25%的概率正常。
面对这样的遗传风险,有没有办法从源头阻断疾病传递?
有办法!——第三代试管婴儿技术(PGT-M)提供了一个可行的答案。
什么是第三代试管婴儿技术(PGT-M)?
中信湘雅生殖与遗传专科医院遗传中心罗俊杰医生表示:简单说,就是精子和卵子在体外受精形成胚胎后,在移植前取一小部分细胞进行遗传学检测,提前判断哪些胚胎是“不合格”的(即患病胚胎),只选择基因正常的胚胎进行移植。
截至2026年3月,中信湘雅生殖与遗传专科医院已出生PGT(即:“三代试管婴儿”技术)婴儿数16314个,正在妊娠中的周期数量是1174个。
适用人群有哪些?
肝豆状核变性携带者夫妻;
其他隐性遗传病携带者夫妻。
如果已经自然怀孕,也可以考虑在孕18周行产前诊断明确胎儿基因型。

肝豆状核变性作为一种常染色体隐性遗传病,虽然会给家庭带来沉重的身心负担,但也是当今三代试管婴儿技术(PGT-M)适用且成熟的典型病种。对于携带ATP7B致病基因的夫妇,完全有机会生育一个健康孩子。
PGT-M技术打破了家族遗传的恶性循环,PGT-M技术不仅赋予了患病家庭生育权,更给了他们“选择不将疾病遗传下去”的希望。
湖南医聊特约作者:中信湘雅生殖与遗传专科医院 遗传中心 罗俊杰
关注@湖南医聊,获取更多健康科普资讯!
(编辑YT)
- 评论