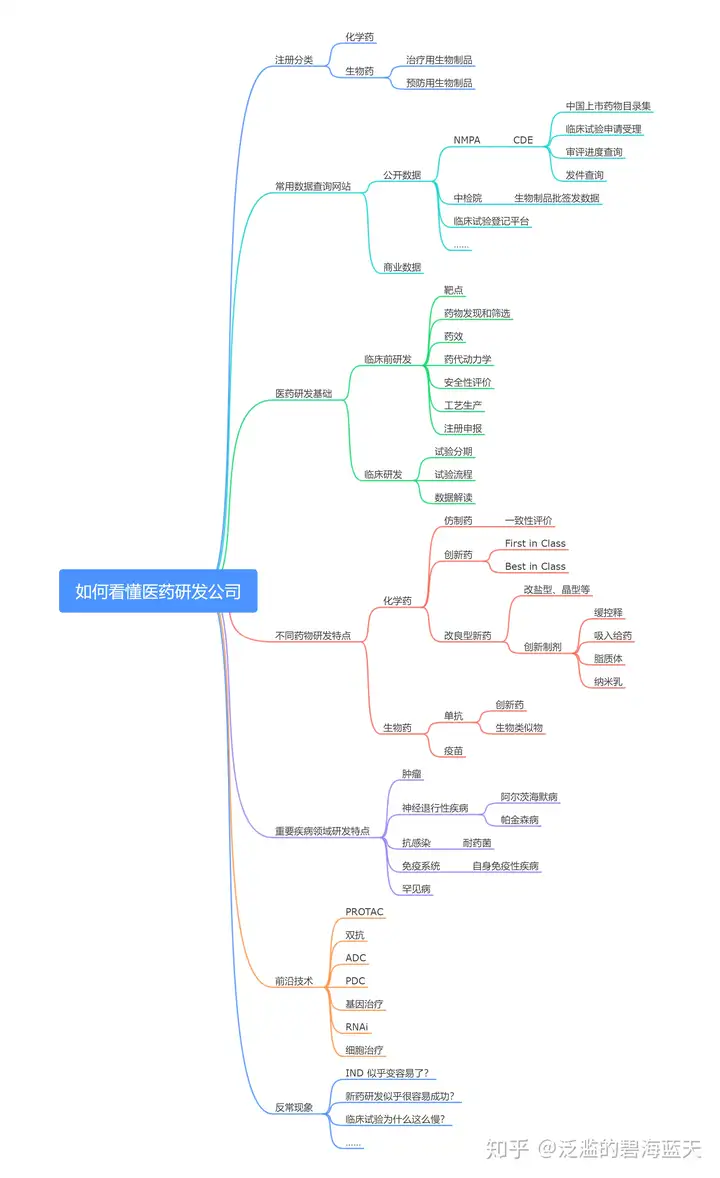
我们平时在读医药公司研发进展新闻的时候,最常见到的就是“某某 1 类新药递交 IND/NDA/BLA”等等。这里就涉及到了药品注册申请和注册分类的内容,作为《看懂医药研发公司》系列文章的第一篇,我们还是先把注册分类搞清楚,从注册分类的角度可以在一定程度上了解产品的创新程度。

了解药品注册分类,读这篇文章就够了
0
评论 ·
3275
浏览 ·
0
收藏
时间:2022年4月10日 来源:知乎
我们平时在读医药公司研发进展新闻的时候,最常见到的就是“某某 1 类新药递交 IND/NDA/BLA”等等。这里就涉及到了药品注册申请和注册分类的内容,作为《看懂医药研发公司》系列文章的第一篇,我们还是先把注册分类搞清楚,从注册分类的角度可以在一定程度上了解产品的创新程度。
什么是药品注册
《药品注册管理办法》里对药品注册的定义如下:
药品注册,是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出申请,药品监督管理部门对拟上市药品的安全性、有效性、质量可控性等进行审查,作出行政许可决定的过程。
药品注册申请包括药物临床试验申请,药品上市许可申请、上市后补充申请及再注册申请。
简而言之,药品注册就是申请人(现阶段主要是指医药企业)向国家药品监督管理局(NMPA)提出药品注册申请,NMPA 审查后给出药品行还是不行的一个过程。
这里比较重要的地方就是,NMPA 在审评过程中重点关注的是药品的安全性、有效性和质量可控性。医药研发的各项工作,其实就是向 NMPA 证明药品安全、有效、质量可控的过程。大家在去关注某个公司的药品最后能否顺利上市的时候,也只需要去重点关注药品的这三个方面就可以了。当然,具体如何去从这三个方面进行评估,我们在后面的文章里还会再展开讨论,这里就不再赘述。
新修订的《药品注册管理办法》对药品注册申请分类进行了重新调整,调整后比较重要的就是临床试验申请(IND)、药品上市许可申请(NDA/BLA/ANDA)。
药品注册分类
当前国内的药品主要分为以下几类:
化学药品:小分子药物;
治疗用生物制品:治疗用途的大分子;
预防用生物制品:主要就是疫苗;
中药、天然药物:中药材、中药提取物、中成药等等。

根据药品类型不同,注册分类也相应有所调整。这里我们按照新修订的《药品注册管理办法》进行讨论,有兴趣的朋友可以自行去了解一下旧的注册分类。
化学药品注册分类
目前化学药品新注册分类共分为 5 个类别,分别如下:
1 类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。
1 类新药最核心的要求就是全新的分子结构且境内外均未上市。比如恒瑞医药用于治疗 Her2 阳性乳腺癌复发或转移性乳腺癌的吡咯替尼、豪森医药的第三代 EGFR 抑制剂奥美替尼等等都属于化药 1 类新药。
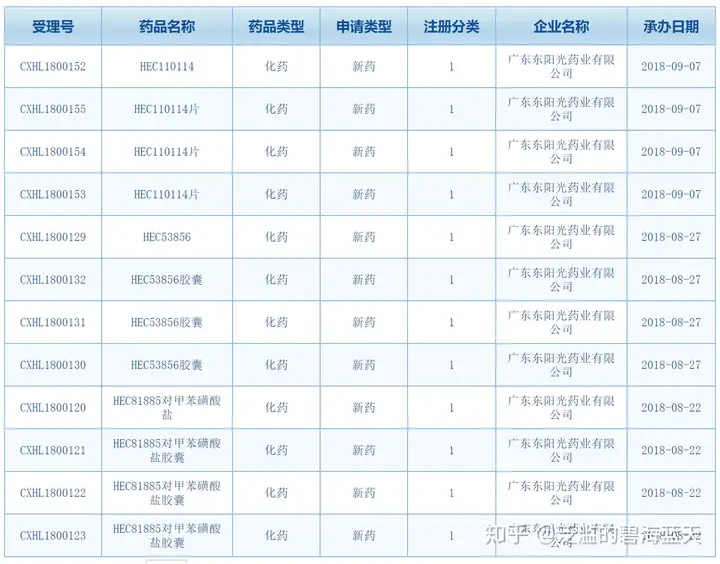
2 类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。
按照定义来看,化药 2 类也是新药范畴,即与 1 类新药一样都要示境内外均未上市。区别在于 2 类新药的活性成份是已知的药物分子,但是在结构、剂型、处方工艺、给药途径、适应症等有所优化,并体现出明显临床优势。下面我们分别举例说明。
结构优化:这里的结构优化主要是指光学异构体优化、成盐或改变酸、碱根、金属元素,或者形成非共价健衍生物。比如说已上市抗生素氧氟沙星是消旋体,后来又推出了光学纯的左氧氟沙星上市,左氧氟沙星的杀菌效果明显优于氧氟沙星。随着药企的专利意识和研发投入的提升,想通过这一途径来取得药品上市越来越难了。
剂型优化:主要指含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径。比如我们硝苯地平最早由德国拜耳在 1969 年开发上市,后来拜耳又推出了硝苯地平控释片(拜新同),拜新同即可归为剂型优化类型。再比如绿叶制药的紫杉醇脂质体。
处方优化:含有已知活性成份的新复方制剂。这个字面比较容易理解,在实际操作中要能找已知活性药物的组合并且体现出复方优势还是比较困难的。2018 年 1 月份,葛兰素史克用于治疗 HIV 的单片复方制剂绥美凯(Triumeq)在国内上市。绥美凯即是包含 Tivicay、Abacavir、Lamivudine 三种药物分子的复方制剂。因其优良的疗效,该药在全球上市一年后就成为“重磅炸弹”药物。
适应症:主要指含有已知活性成份的新适应症的制剂。我们通常说的“老药新用”,就是在说这一路径。最著名的例子莫过于降糖神药二甲双胍了,当前针对该药的新适应症仍在开发中。很多在早期开发阶段先选择某一个适应症争取尽快上市,然后上市后再去拓展适应症的开发策略,也是基于这一路径。这种方式在肿瘤药的开发中可能会最常碰到。
3 类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。
原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。
4 类:境内申请人仿制已在境内上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。
化药 3 类和 4 类区别在于 3 类是境外上市的原研药品,4 类是境内上市的原研药品。将境内外原研区分开,主要应该是考虑到人种差异因素,化药 3 类在临床试验时需要做临床试验验证有效性。
常见的首仿概念,原来还可以简单地以境外原研上市境内无产品上市来界定。随着国内原研产品的不断上市,首仿就只能是看是不是国内首家仿制药上市了。
5 类:境外上市的药品申请在境内上市。
这个主要针对境外上市的药品。
生物制品注册分类
生物制品主要包括疫苗、血液制品、生物技术药物、微生态制剂、免疫调节剂、诊断制品等,根据用途可以分为预防用生物制品和治疗用生物制品两类。
预防用生物制品分类
按照产品成熟度不同,预防用生物制品可以分为 5 个类别。
1 类:新型疫苗:指境内外均未上市的创新疫苗。在境内外已上市制品基础上制备的新的结合疫苗或者联合疫苗,与境内外已上市疫苗对应的抗原群或者型别不同的疫苗,境内外已上市疫苗保护性抗原谱不同的重组疫苗,更换其他未经批准使用过的表达体系或者细胞基质生产的疫苗,DNA疫苗,应当按照注册分类1类申报。
康希诺的重组埃博拉病毒疫苗、康泰生物的重组肠道病毒71型疫苗(汉逊酵母)、智飞龙科马的四价重组诺如病毒疫苗(毕赤酵母)等等都是按 1 类疫苗申报的。
2类:改良型疫苗:指对境内已上市疫苗产品进行改良创新,使新产品具有重大技术进步和/或具有显著临床优势,或者对制品的安全性、质量控制方面有显著改进的疫苗。包括:
2.1 疫苗实体的改变,例如灭活疫苗或减毒活疫苗已上市,申报基因重组疫苗;减毒活疫苗已上市申报灭活疫苗等;
2.2 基于重大技术改进的疫苗,包括疫苗菌毒种/生产工艺/制剂处分等的改进。如,由非纯化或全细胞(细菌、病毒等)疫苗改为纯化或者组份疫苗等;采用新的菌毒株、细胞基质或表达体系的疫苗;改变已上市结合疫苗的载体;改变灭活剂(方法)或者脱毒剂(方法),采用新工艺制备并且实验室研究资料证明产品安全性和有效性明显提高的疫苗;
2.3 改变佐剂或采用新佐剂的疫苗;
2.4 改变给药途径或改变剂型,且新的给药途径或剂型具有显著临床意义;
2.5 改变免疫剂量和免疫程序,且新免疫剂量和免疫程序具有显著临床意义;
2.6 改变适用人群,且新适用人群具有显著临床意义;
3 类:境外上市、境内未上市的疫苗。
4 类:境内已上市的疫苗。
5类:进口疫苗:根据其成熟程度分为上述同样4种情形。
5.1 新型疫苗;
5.2 改良型疫苗;若在境外已上市制品基础上进行改变的,应当按照注册分类2类申报。
5.3 境外上市、境内未上市的疫苗;
5.4 境内已上市的疫苗。
后面分类可以参照化药分类的解释进行理解,这里也不再赘述。
治疗用生物制品
治疗用生物制品也可以分为 5 个类别。
1 类:新型生物制品:指未在境内外上市的全新治疗用生物制品。由已上市的治疗用生物制品成分组成的新复方制剂,在境内外已上市制品基础上,改变氨基酸序列、改变蛋白质高级结构和多聚体形态的,改变翻译后修饰,或者对产物进行化学修饰(包括PEG化偶联修饰等)的,应当按照注册分类1类申报。
全新的基因治疗和细胞治疗类生物制品(例如创新机理、新载体、新靶细胞等),应当按照注册分类1类申报。
国内君实、信达、恒瑞、百济的 PD-1 抗体都是按 1 类新型生物制品申报的。
2 类:改良型生物制品:指在境内外已上市制品基础上,对其制剂水平的结构(如影响释放和生物利用度的粒径及其分布、包合、聚合结晶等制剂技术产生的结构改变)剂型、处方工艺、给药途径等进行优化,对适应症进行增加、优化或者用药人群的增加(如增加儿童、老年人用药人群);或者首次采用DNA重组技术制备的制品(例如以重组技术替代合成技术、生物组织提取技术等)、与境内外已上市制品制备方法不同的制品(例如采用不同表达体系、宿主细胞等)。
在境内外已上市制品基础上进行改进的基因治疗和细胞治疗类生物制品,应当按照注册分类2类申报。
除了儿童用药的外推之外,改良型新生物制品应当具有明显临床优势,或者对制品的安全性、质量控制方面有显著的改进。
改良型生物制品的逻辑和改良型化药也类似,可以对照理解。
3 类:境外上市、境内未上市的生物制品:若原研药/参照药仅在境外上市,申请人按生物类似药研发的生物制品可按此类别申报临床试验申请;不能按生物类似药技术要求进行研制申报的,申请人应根据制品情况按照注册分类 1 类或者 2 类申报临床试验申请。
原则上,注册分类 3 的生物制品应当在其原研药/参照药获得境内临床试验批准后方可开展临床试验。完成临床试验后,根据当时情势按照适宜的注册分类提交上市申请。
4 类:境内已上市的生物制品。包括:
4.1 生物类似药;
4.2 不能按生物类似药技术要求进行研制申报的生物制品。
这里提到了生物类似药的概念。通俗地理解,可以说生物类似药就是生物仿制药。但由于生物大分子的特殊性,不同的生产工艺生产出来的相同序列的生物大分子,也可能会在临床上表现出不同疗效和安全性。因此,生物制品的仿制被叫作生物类似药,相应地,在临床试验阶段,也比化学仿制药的临床投入大得多。
5 类:进口生物制品:根据其成熟程序分为上述同样4种情形。
5.1 新型生物制品;
5.2 改良型生物制品;
5.3 境外上市、境内未上市的生物制品:包括境外已上市的原研药提交的临床试验申请和上市申请,以及按生物类似药研发的进口生物制品的临床试验申请按此类申报。不能按生物类似药技术要求进行研制申报的,申请人应根据制品情况按照注册分类5.1类或者5.2类申报临床试验申请;
5.4 境内已上市的生物制品。若原研药已在境内上市,申请人按生物类似药研发的生物制品按此类申报。
中药、天然药物注册分类
中药和天然药物的区别在于中药是在我国中医药理论指导下使用,天然药物是在现代医药理论指导下使用。中药、天然药物注册分类也分为 5 类。
1 类:创新药。指含有未在中药或天然药物国家标准的【处方】中收载的新处方,且具有临床价值的药品,包括单方制剂和复方制剂。
2 类:改良型新药。指对已上市销售中药、天然药物的剂型、给药途径、适应症等进行优化,且具有明显临床优势的药品。
3 类:古代经典名方。指目前仍广泛应用、疗效确切、具有明显特色与优势的清代及清代以前医籍所记载的方剂。
4 类:同方类似药。指处方、剂型、日用生药量与已上市销售中药或天然药物相同,且在质量、安全性和有效性方面与该中药或天然药物具有相似性的药品。
5 类:进口药。指境外上市的中药、天然药物申请在境内上市。
近年来,审评中心对中药、天然药物申请的受理数量下降非常明显。相比于美国 FDA 在植物药方面的注册要求,国内在天然来源的药物审评方面还有待提升。我们开篇就已经提到,药物能否被批准上市,需要从安全、有效、质量可控三个方面进行把控。无论是中药还是天然药物,都应该在符合安全、有效、质量可控的基本要求下,再来探讨理论层次的问题。
小结
本篇简单介绍化学药、生物制品和中药、天然药物的注册分类,主要是为了让大家能有个大致的印象。概括来说,注册分类主要需要把握以下几点:
1 类创新药:研发难度较大,上市成功率较低,研发投入也大,研发周期平均在 10 年左右,一旦成功,将带来丰厚的回报。现在看来,国内企业对于创新药的预期相对还是偏乐观,对风险的考虑不足。历史原因再加上国内的行业环境,国内医药企业研发很少会主动宣告临床研发失败。我们在实际评估时,需要特别关注企业重要产品的临床研发进度。从逻辑上讲,如果临床进度落后于预期太多,这里面大概率是存在没有批露的问题的。
2 类改良型新药:大多是原研厂家为了延长自己产品的生命周期推出的新一代产品。研发难度相对较低,上市成功概率较大。评估的核心在于是否具有临床优势,做得好可以成神。
3 类仿制药:化药仿制药,“4 + 7”背景下,化药仿制药的逻辑除了抢首仿外,成本优势可以说是生死攸关。控制成本的前提下,企业规模和原料药优势就愈发重要。未来,仿制药厂和原料药厂的并购可能会成为常态。生物类似物,因为还有较高的研发壁垒,成功上市后,短期内价格应该也不会像化药仿制药一样进入肉搏战。企业凭借领先的研发效率,还是可以获得相对长时间的竞争优势。
- 评论
发布
更多